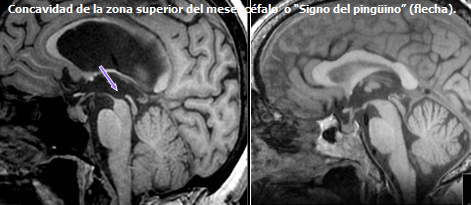
Williams, David and Lees, Andrew. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol 2009; 8: 270–79.
Hoy les traigo este trabajo de Agosta y col, una comparación entre los dos subtipos más frecuentes de Parálisis Supranuclear progresiva:
La PSP puede
ocurrir con 2 presentaciones clínicas principales (Williams et al., 2005). Más
50% de los casos muestran inestabilidad postural de aparición temprana y
caídas, parálisis mirada vertical supranuclear, disfunción cognitiva y se
clasifican como síndrome de Richardson (PSP-SR) (Williams et al., 2005). El
otro gran grupo está entre el 20% de los casos y son clasificados como
PSP-parkinsonismo (PSP-P), que se caracteriza por la aparición asimétrica,
temblores, rigidez, moderada respuesta inicial a la levodopa (Ldopa), una edad
de inicio de la enfermedad más tardío, y un curso de la enfermedad más
favorable en comparación con PSP-SR (Williams et al., 2005). En casos de PSP-P,
la distinción de la enfermedad de Parkinson (PD) puede ser imposible a principios
de la enfermedad (Williams y Lías, 2010; Williams et al., 2005). Sin embargo,
la presencia de alucinaciones visuales, disquinesias tardía inducida por drogas
y disfunción autonómica final han resultado ser muy poco frecuente en PSP-P y
pueden ayudar a distinguir entre PSP-P y EP y en un PSP-P cuando se presentan
es en una etapa tardía en el curso de la enfermedad (Williams y Lías, 2010;
Williams et al., 2005). Hasta que los estudios demostraron que esa deposición
de tau (Jellinger, 2008; Morris et al., 2002; Williams et al., 2007a), así como
gris materia (GM) y atrofia de materia blanca (WM) (Schofield et al, 2011) son
significativamente más extensos y graves en PSP-RS que en casos de PSP-P.
El índice de
parkinsonismo por resonancia magnética (IPRM o MRPI), que combina las
mediciones de áreas de mesencéfalo y puente, así como grosor promedio del
Pedúnculos cerebelosos superiores (PCS) y los Pédunculos cerebelosos medios
(PCM), ha demostrado ser una medida precisa para el diagnóstico diferencial
entre PSP-RS y EP o la variante de Parkinson de atrofia de sistema múltiples (AMS-P)
(Quattrone et al., 2008). Las imágenes por resonancia magnética de tipo Volumétrica
también pueden ser útiles para distinguir subtipos de PSP (Agosta et al., 2010;
Longoni et al, 2011). PSP-SR resultó estar asociada con una mayor atrofia
cerebral en comparación con PSP-P, que fue más severo en el PCS (Agosta et al.,
2010; Longoni et al, 2011), pedúnculos cerebrales y a lo largo de los tractos piramidal
(TCE) (Agosta et al., 2010). La degeneración del tracto es, probablemente uno
de los principales contribuyentes a las diferentes presentaciones clínicas de
PSP. La RM por Difusión el efecto de la microestructura de los tejidos sobre el movimiento de traslación al azar de las moléculas de agua en los tejidos biológicos (Basser y Pierpaoli, 1996). Una caracterización completa de difusión puede ser proporcionada por el método basado en el tensor de difusión (TD).
RM TD tiene el potencial para revelar lesiones que pueden no ser evidentes con otras modalidades (Basser y Pierpaoli, 1996). Sin embargo, la mayoría de los estudios de resonancia magnética DT realizado hasta ahora no incluye las presentaciones clínicas atípicas de PSP. En este estudio, hemos querido utilizar RM TD para investigar en vivo similitudes y diferencias de la distribución regional de los tractos daños en PSP -SR y PSP-P, comparar estos resultados con los correspondientes patrones de atrofia cerebral y cuantificar la capacidad de RM de TD en diferenciar los síndromes PSP a nivel individual, cuando se utiliza en combinación con el IPRM. Tenemos la hipótesis de que las las anormalidades microestructurales puede ser más grave y generalizada en PSP-SR, especialmente las que implican los pedúnculos cerebelo y región frontal del cerebro. También establecer la precisión de diagnóstico en diferenciar cada síndrome PSP de personas ancianas sanas y mutuamente entre los subtipos.
Discussion
Este estudio evaluó en vivo la distribución regional de anormalidades cerebrales microestructural y volumétrica en Parálisis supranuclear progresiva - Síndrome de Richarson (PSP-SR) y Parálisis supranuclear progresiva -Parkinsonismo (PSP-P). Todos los pacientes PSP mostraban anormalidades de difusividad en el cuerpo calloso, frontoparietal y conexiones frontotemporo-occipital. Por el contrario, las regiones de núcleos y infratentorial fueron severamente afectadas en casos de PSP-RS y relativamente escatimadas en aquellos con PSP-P.
La PSP-SR está asociado con un patrón más severo de atrofia en comparación con PSP-P. Este estudio también muestra que un modelo estadístico basado en la MRPI así como las variables de MRI DT capaz de capturar toda la extensión de las anomalías patológicas visibles, es más preciso que la volumetría infratentorial solo en diferenciar pacientes entre las 2 variantes de PSP.
La PSP mostró un patrón generalizado de anormalidades en difusividad y FA que afectan a la mayoría de los tractos cerebral, bilateralmente.
Las alteraciones en RM TD se observaron en tractos situados en:
Los lóbulos frontales, Cuerpo calloso en su porción anterior, cíngulo y fasciculo longitudinal superior (FLS) y en aquellos que pasan por los lóbulos temporales, tales como el fascículo uncinados, FLI y FOFI. Junto con atrofia cortical frontal y de núcleos (ganglios) basales (Agosta et al., 2010; Cordato et al., 2005; Josephs et al, 2011), daños a las redes de sustancia blanca frontotemporal y frontoparietal tienen probabilidades de ser responsable por el desarrollo de la típica disfunción ejecutiva y síntomas conductuales en PSP. En ambos grupos de pacientes, anomalías difusividad y de la FA también involucraron el TCE bilateralmente, desde la cápsula interna hasta regioners en estrecha vecindad con las áreas motoras y premotor. Se sabe que el TCE puede estar involucrado en la PSP y de hecho un tercio de los pacientes muestran signos piramidales, la diferencia más notable en la medida y distribución de daños WM entre los 2 subtipos PSP fue encontrado en las estructuras infratentorial y de los núcleos Talámicos.
PSP-SR mostraron un daño severo al: mesencéfalo, pedúnculos cerebeloso, núcleos y sustancia blanca del cerebelo y radiaciones talámicas. Por el contrario, tales estructuras estan mejor preservadas en PSP-P. Además, PSP-RS se asoció con una mayor pérdida de sustancia blanca, incluyendo el mesencéfalo, PCS y tanto de las regiones y sus inmediaciones de Corteza motora y premotora. Los hallazgos de resonancia magnética de Tensor de Difusión (TD) observaron en los dos subtipos de PSP que probablemente reflejan la distinta disfunción tau con la participación de los microtúbulos en los axones.
Estudios patológicos mostraron que los pacientes de PSP-P tienen una patología tau menos grave que aquellos con PSP-RS, particularmente en el núcleo dentado cerebeloso y la sustancia blanca del cerebelo, corteza cerebral, puente y núcleo caudado. La composición ultraestructural de la maraña-tau insoluble también difiere entre los 2 grupos: isoformas tau 4-repetir en las inclusiones neuronales y gliales son predominantes en pacientes con PSP-RS, mientras que una mayor contribución de isoformas tau 3-repetición a la fracción insoluble-tau total ha sido descrita en PSP-P.
La Atrofia cerebral estaba más extendida en PSP-RS que pacientes de PSP-P y puede ser un factor que contribuye a la mayor gravedad de los daños a la sustancia blanca en los tractos que conducen vistos en el primer grupo de pacientes. La relativamente leve severidad de los daños en PSP-P, así como su patrón menos generalizado, están probablemente entre las razones de las diferencias clínicas que distinguen a este síndrome de PSP-RS, incluyendo su mayor duración de la enfermedad y mejor pronóstico. Por ejemplo, el temblor con una moderada capacidad de respuesta de l-dopa, que caracterizan los casos PSP-P en la fase temprana de la enfermedad, pueden ser debido a un agotamiento menos severo de la dopamina en el mesencéfalo, de estos pacientes.
Los daños a la red de control motor que incluye el cerebelo, mesencéfalo, núcleos basales, tálamo y corteza podría explicar la mayor constelación de déficit motor funcional en PSP-RS. En el PCS, sus fibras unas se proyectaran hacia la corteza a través del haz dento rubro tálamo cortical otras ejerceran también inadecuadamente hacia los núcleos reticular y vestibular del tronco encefálico, que puedan estar involucrados en el déficit de equilibrio y la postura de los pacientes con PSP-RS. Recientemente, se ha encontrado una atrofia talámica mucho más grave relacionados con la duración de la parálisis de mirada supranuclear en casos con PSP patológicamente demostrados.

Sus conclusiones:
Hemos encontrado que el IPRM es capaz de diferenciar PSP-RS de controles con una alta precisión de diagnóstico (92%). Sin embargo, cuando la IPRM se utilizó para distinguir PSP-P de controles, la capacidad de diagnóstico fue subóptimo (70%). También que IPRM era sólo moderadamente preciso en diferenciar las 2 variantes PSP (precisión 77%). Por el contrario, cuando el IPRM se combinó con la evaluación global de la PSP relacionados con el patrón de anomalías mediante RM-TD, la capacidad de diferenciar PSP-P de controles y PSP-RS mejora a un 141% y 96%, respectivamente.
Agosta F, Pievani M, Svetel M, Ječmenica Lukić M, Copetti M, Tomić A, Scarale A, Longoni G, Comi G, Kostić VS, Filippi M. Diffusion tensor MRI contributes to differentiate Richardson's syndrome from PSP-parkinsonism. Neurobiol Aging. 2012 Mar 12. [Epub ahead of print]