Repasemos brevemente entonces estos casos:
El Síndrome de Guillain-Barré. (SGB) Wardrop y Ollivier, tal vez son precursores y se sabe que lo publican en 1834, luego la descripción de un cuadro ascendente por Landry en 1859, la mejor descripción por Guillain, Barré y Strolh en 1916 de una polineuritis benigna con disociación albumino citológica en el LCR. Asbury, Arnason y Adams en 1969, establecieron que la lesión esencial desde el inicio es una lesión inflamatoria mononuclear perivascular de las raíces y el nervio.

Es una polineuropatia aguda que se produce en todas partes del mundo y en todas las estaciones, afecta niños y adultos y no tiene predominio franco de género, En general una infección respiratoria o intestinal precede al cuadro neuropático entre 1 a 3 semanas en alrrededor del 60 % de los casos Presenta un inicio súbito con parálisis motora ascendente, parálisis bulbares o respiratorias y pocos síntomas sensoriales, sin afectación esfinteriana, con alteraciones vasculares autonómicas; y no hay nivel sensitivo.
Las manifestaciones clínicas son el resultado de una reacción inmunológica mediada por células dirigidas contra los nervios periféricos. Se identifican diversos autoanticuerpos, el más prominente de los cuales es el anti-GQ1b, que se halla en casi todos los pacientes con oftalmoplejía .
Variantes del patrón típico del SGB.
En ocasiones se afectan primero los músculos faríngeos, cervicales, y braquiales, lo que dificulta la deglución y debilita el cuello y la porción proximal del brazo (Ropper, en 1986). Pero al que hacemos mención hoy es al Síndrome de Miller Fisher, o simplemente Síndrome de Fisher (SF) para evitar confusión de que se trate de 2 personas distintas, el cuadro consiste en Oftalmoplejía completa, Ataxia y Arrefexia. La debilidad se reporta en un 30%. Se presenta entre el 5 y 10% de los casos. Su incidencia anual es de 0.09 por 100,000 habitantes. Afecta más a hombres que a mujeres con una relación 2:1.

La edad promedio de aparición es a los 40 años con un intervalo de 13 a 78 años. Se encuentra con frecuencia el antecedente de infección por campilobacter jejuni, hemofilus influenzae, citomegalovirus y micoplasma pneunioniae, entre otros. El cuadro clínico se desarrolla entre los 10 a 14 días después de la infección. La etiopatogenia se basa en un fenómeno de mimetismo molecular en relación a los gangliósidos GQ1b, GD3 y GT1a, presentes en la mielina de los nervios periféricos y en las áreas paranodales de los nervios craneales III,IV,V y VI. Aunque la afección distinta a los otros nervios craneales se encuentra en el 40 al 57%. Parálisis facial en el 35% y la afección bulbar en el 16%. La debilidad muscular fue significativa en el 25% y la alteración sensitiva en el 52%. En más del 90% de los pacientes con SMF se detectan anticuerpos anti-GQ1b y los títulos de IgG son mayores al inicio del cuadro.
La edad promedio de aparición es a los 40 años con un intervalo de 13 a 78 años. Se encuentra con frecuencia el antecedente de infección por campilobacter jejuni, hemofilus influenzae, citomegalovirus y micoplasma pneunioniae, entre otros. El cuadro clínico se desarrolla entre los 10 a 14 días después de la infección. La etiopatogenia se basa en un fenómeno de mimetismo molecular en relación a los gangliósidos GQ1b, GD3 y GT1a, presentes en la mielina de los nervios periféricos y en las áreas paranodales de los nervios craneales III,IV,V y VI. Aunque la afección distinta a los otros nervios craneales se encuentra en el 40 al 57%. Parálisis facial en el 35% y la afección bulbar en el 16%. La debilidad muscular fue significativa en el 25% y la alteración sensitiva en el 52%. En más del 90% de los pacientes con SMF se detectan anticuerpos anti-GQ1b y los títulos de IgG son mayores al inicio del cuadro.
Por otra parte Bickerstaff y Cloake en 1951 describieron a 3 pacientes titulando a su trabajo romboencefalitis y mesencefalitis. Aquí podemos resaltar que desde el punto de vista embriogénico el rombo encéfalo o cerebro posterior está constituido por el puente, médula oblonga, cerebelo y el cuarto ventrículo, y entre el prosencéfalo y el rombo encéfalo se encuentra el mesencéfalo o cerebro medio, conformado por Pie mesencefálico, el tegmentun mesencefálico el tectum mesencefálico y el acueducto cerebral.
Bickerstaff publicó primero que Fischer, pero probablemente es menos conocido, por su menor frecuencia de aparición, y pocas evidencias patológicas por la relativa forma benigna en la evolución de sus casos, pero debe quedar claro que existen casos que progresan a muerte.
Posteriormente con la publicación de 8 casos en 1956, Bickerstaff plantea la denominación de encefalitis del tallo cerebral y lo caracteriza clínicamente la presencia de: Oftalmoplejía, Ataxia, Somnolencia de inicio que puede alcanzar el coma, y la presencia de signos de afección del tracto piramidal como hiperreflexia y signo de babinski, ausencia de cutáneos abdominales más aun si hay evidencia de contracción de musculatura abdominal y movimientos fuertes de las costillas inferiores.
La encefalitis troncoencefálica de Bickerstaff (ETB o BBE) es una rara enfermedad neurológica post-infecciosa caracterizada por la asociación de oftalmoplejía externa, ataxia y alteración de la conciencia (somnolencia, estupor o coma) o hiperreflexia. Se desconoce su prevalencia. Los pacientes suelen presentar diplopía y alteración de la marcha después de infecciones de las vías respiratorias o gastrointestinales. La oftalmoplejía externa es progresiva (a lo largo de las 4 semanas de aparición) y relativamente simétrica. También puede observarse tetraparesia simétrica flácida en más de la mitad de los pacientes, junto con una discapacidad sensorial superficial o profunda, debilidad facial, parálisis bulbar, oftalmoplejía interna, blefaroptosis y nistagmo. En la fase aguda de la enfermedad, la BBE puede ser muy grave, produciéndose oftalmoplejía completa, diplejía facial y parálisis completa de manos y piernas, lo que se asemeja a una muerte cerebral. La aparición de la BBE ha sido descrita tras la infección por diferentes agentes, incluyendo los citomegalovirus, Campylobacter jejuni y Mycoplasma pneumonie. Aunque no se conoce exactamente el mecanismo patológico de la enfermedad, la BBE está asociada con la presencia de anticuerpos antigangliósidos, anti-GQ1b. Estudios inmunohistoquimicos revelan gran afinidad de estos anticuerpos en región oculomotor, mucho menor en núcleos cerebelosos profundos, la sustancia gris de tallo cerebral, médula espinal y células del ganglio dorsal y huso muscular. La afección del ganglio dorsal puede explicar la arreflexia y la ataxia.
El diagnóstico se basa en los hallazgos clínicos, historial del paciente, análisis del líquido cefalorraquídeo (que muestra un aumento de los niveles de proteínas), detección de anticuerpos IgG anti-GQ1b (no presentes en todos los pacientes), estudios de RM (que revelan anomalías en la fosa posterior, en la sustancia blanca o en el tálamo) y exámenes neurofisiológicos (electroencefalograma y electromiografía, útiles para valorar la afectación del sistema nervioso central, en particular la afectación axonal). La BBE se solapa clínicamente con el SF, una variante del síndrome de Guillain-Barré que afecta a los nervios craneales, y con las formas axonales del GBS (la neuropatía axonal motora aguda y la neuropatía axonal aguda motora y sensitiva, véase estos términos) en pacientes con debilidad en las extremidades. Diversos autores han sugerido que la BBE, el MFS y el GBS representan manifestaciones variables del mismo espectro clínico. EL diagnóstico diferencial en pacientes con BBE también debe incluir la encefalomielitis diseminada aguda (ADEM), así como enfermedades neurológicas postinfecciosas poco frecuentes. El manejo se basa en uso de corticoesteroides, inmunoterapia con inmunoglobulina intravenosa (IgIV) o con plasmaféresis. Aunque el cuadro clínico es grave, la enfermedad suele seguir un curso monofásico con una remisión completa de los síntomas a los 6 meses en más de la mitad de los pacientes. Otros pacientes pueden tener signos residuales de leves a graves.
En una serie del año 2003 de Odaka et al. 16% de los pacientes requirió ventilación asistida. A los seis meses el 66% está en completa remisión. En los pacientes sin debilidad en los miembros se observaron secuelas motoras o sensitivas en 14% y diplopía o ataxia en 9%. Hubo 3 defunciones que representa un 5%, debida a complicaciones de la enfermedad.
La edad tenía una mediana de 39 años, con predominio de los hombres (3:2). La cuadriparesia fláccida simétrica en el 60% y se consideró como un SGB superpuesto. El 92% tenía el antecedente de una enfermedad infecciosa reciente. La alteración de la conciencia fue frecuente (74%) y el signo de Babinski en el 40 % de los casos. La diplejía facial se observó en el 45 % y las anormalidades pupilares y la parálisis bulbar en el 34%. Los reflejos miotáticos estuvieron disminuidos o ausentes en el 58% , normales en el 8% y aumentados en 34%.
Los anticuerpos IgG anti-GQ1b séricos fueron positivos en el 66%. Usando métodos de inmunohistoquímica, Chiba et al demostraron la singular distribución del epítope GQ1b en las regiones paranodales de los nervios motores oculares humanos, lo cual sugiere una estrecha relación entre anticuerpos anti-GQ1b y oftalmoplejía.
Los anticuerpos IgG anti-GQ1b séricos fueron positivos en el 66%. Usando métodos de inmunohistoquímica, Chiba et al demostraron la singular distribución del epítope GQ1b en las regiones paranodales de los nervios motores oculares humanos, lo cual sugiere una estrecha relación entre anticuerpos anti-GQ1b y oftalmoplejía.
La RM mostró en el 30 por ciento lesiones hiperintensas en la fosa posterior, la sustancia blanca cerebral, o el tálamo. Las imágenes de Resonancia Magnética ponderadas por difusión (DWI) Diffusion Weighted Imaging, han revelado señales hiperintensas en el T2 que están asociadas con valores altos del coeficiente de difusión aparente (ADC) Apparent Diffusion Coefficient, lo que indica un edema vasógeno, más que citotóxico.Las imágenes de la RM de edema vasógeno descendente del tallo cerebral se correlacionan muy bien con las manifestaciones clínicas de la ETB o EBB. Los estudios de seguimiento de RM han demostrado que las lesiones desaparecen completamente.



RM en una mujer de 81 años. Se observa incremento de la señal en mesencéfalo y ponto mesencefálico dorsal, en FLAIR (fluid-attenuated inversion recovery) Roos Raymond et al.
Se observa realce con Gadolineo en T1
Estas imagenes de un paciente que presento en sintesis: Ataxia, diplopia, disartria, disturbio del estado de conciencia, con signo de babinski. La afectacion motora se atribuyo a la afeccion del tracto corticoespinal, la edad no es tipica, en este caso usaron corticoesteroides y la evolucion fue favorable. Publicado por: Raymond P. Roos, MD ; Betty Soliven, MD ; Fernando Goldenberg, MD ; Aamir Badruddin, MD ;Joseph M. Baron, MD. An Elderly Patient With Bickerstaff Brainstem Encephalitis and Transient Episodes of Brainstem Dysfunction. Arch Neurol. 2008;65(6):821-824.
En los pacientes con SF, la RM contrastada muestra realce de los pares craneanos III, IV y VI, sin embargo, en algunos casos puede haber anormalidades en el tallo cerebral en las imágenes de RM.
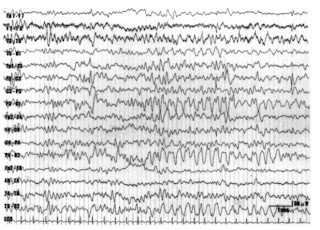
El Electrencefalograma puede reelar la presencia de ondas lentas y en algunos puntas onda.
En los pacientes con SF, la RM contrastada muestra realce de los pares craneanos III, IV y VI, sin embargo, en algunos casos puede haber anormalidades en el tallo cerebral en las imágenes de RM.
El Electrencefalograma puede reelar la presencia de ondas lentas y en algunos puntas onda.
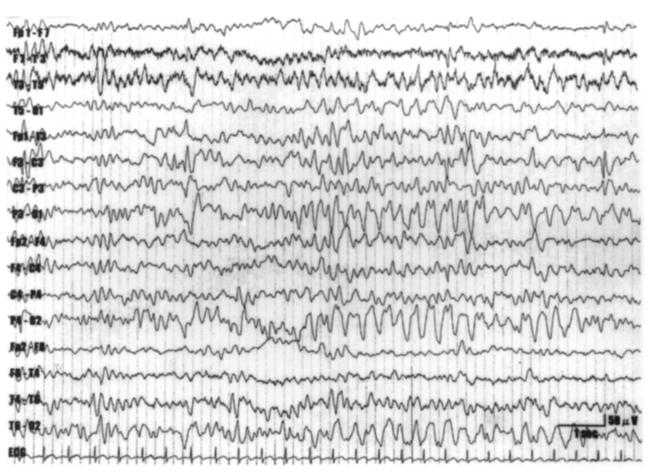
Presencia de puntas en region frontal izquierda (ubicacion central) y ondas lentas de 2 – 3 Hz. predominantemente posteriores en lobulos occipitales.
La autopsia de uno de los pacientes con ETB demostró infiltración linfocítica perivascular con edema y nódulos gliales en el tallo cerebral. Casi todos los pacientes tuvieron un curso monofásico remitente.
No hay comentarios:
Publicar un comentario